Article Text

Abstract
With the projected expansion of the biosimilars market, there will be an increased propensity for the substitution of reference biological products with cheaper biosimilars for economic reasons (ie, non-medical switching). This will lower the cost per patient and should provide the benefit of wider access to biological therapies. However, it is essential that patients and clinicians fully understand the rationale for non-medical switching and its potential implications in terms of efficacy, safety, and immunogenicity. To date, clinical experience supports the use of biosimilars and a growing body of evidence from clinical trials and real world observational studies specifically supports clinical decision making around non-medical switching. Equally, as non-medical switching becomes more common, it is essential that pharmacovigilance systems adapt to handle the increasing volumes of data needed to effectively monitor the use of biosimilars and detect new signals. This will require a reduced reliance on registries, as well as streamlining and integration of existing systems to allow a frequent cycle of online reporting of adverse events by healthcare professionals, analysis by national authorities, and feedback to treating clinicians. This article considers the current use and future uptake of biosimilars from a clinical perspective.
Statistics from Altmetric.com
Considering the growing availability of biosimilars
Biosimilar development aims to improve access for patients while reducing the costs associated with expensive biological treatments. According to a large database of biologics, there are currently more than 1500 biologics on the market or in clinical development; of these, 146 are reference products, 840 are biosimilars, and 515 are ‘biobetters.’1 For inflammatory disease alone, with the imminent patent expiry of several biological (reference) agents, at least 37 biosimilars are in clinical development.2 Given the likelihood of new reference products with novel mechanisms of action, and the availability of multiple biosimilars for each reference product, the number of biologics and complexity of treatment options for inflammatory disease is set to increase substantially. Hence, physicians are faced with the prospect of complex clinical decision making around the appropriate use of biologics and biosimilars.3
Considering switching
To facilitate an optimal clinical delivery of biologics and biosimilars, a requirement exists for harmonisation of the nomenclature around their use. In the following sections, we define switching as the decision to exchange one medicine for another with the same therapeutic intent.4 Interchangeability is a regulatory designation whereby an interchangeable biological product, in addition to meeting the biosimilarity standard, is expected to produce the same clinical result as the reference product in any given patient with no impact on efficacy or safety.4 Finally, automatic substitution refers to the practice of dispensing one medicine instead of another equivalent and interchangeable medicine at the pharmacy level, without consulting the prescriber.4
Medical and non-medical switching
In the context of biological therapies, switching can refer to a change from one reference biologic to an alternative biologic, between a reference biologic and a biosimilar (or vice versa), or between two different biosimilars of the same reference biologic. From a clinical standpoint, each of these options could be used for different reasons and have potentially different consequences. As an aid to understanding the different options, switching can be sub-categorised into medical switching and non-medical switching.
Medical switching, whereby one medication is exchanged for another at the discretion of the prescribing physician, is performed with the goal of optimising treatment benefit, improving convenience, or minimising adverse events.5 In most instances, a different reference biologic is introduced after the first biologic has failed, and patients usually remain on the substitute biologic. Evidence based recommendations to guide clinicians in the medical switching of biologics are available for several conditions, including rheumatoid arthritis6 7 and Crohn’s disease.8 These guidelines recommend that patients who experience an inadequate response or an intolerable adverse event during treatment with a biologic should be switched to a different biological agent.
In contrast, non-medical switching (NMS) is the switching of biologics in patients with well tolerated, adequate therapy.9–11 Broadly, two types of NMS can occur: between different biological agents of the same class, or more commonly from a reference product to its biosimilar or vice versa.10 12 NMS is typically driven by economic reasons (cost savings and/or procurement policies) and may be enforced through guidelines and/or legislation.9–11 13 14 Less commonly, NMS can be driven by patient preference.9 10 Efficacy, safety, or convenience benefits are not expected from NMS; however, a prescribing physician should be part of the decision and incentives may exist to switch their patients. Where NMS occurs without the involvement of the prescribing physician, this can be considered an automatic substitution. Currently there are no formal recommendations to guide NMS, representing an important unmet need. As discussed below, clinical evidence supports NMS between reference biologic and biosimilar. However, if an NMS decision requires the substitution of one biosimilar for another biosimilar, there is a lack of clinical evidence for physicians to refer to because a biosimilar may not be evaluated against more than one reference for the purpose of regulatory approval.15 16 Consequently, comparability studies are generally performed between a biosimilar and its reference product but not between one biosimilar and another (figure 1). Therefore, NMS between different biosimilars is not desirable from a clinical perspective.
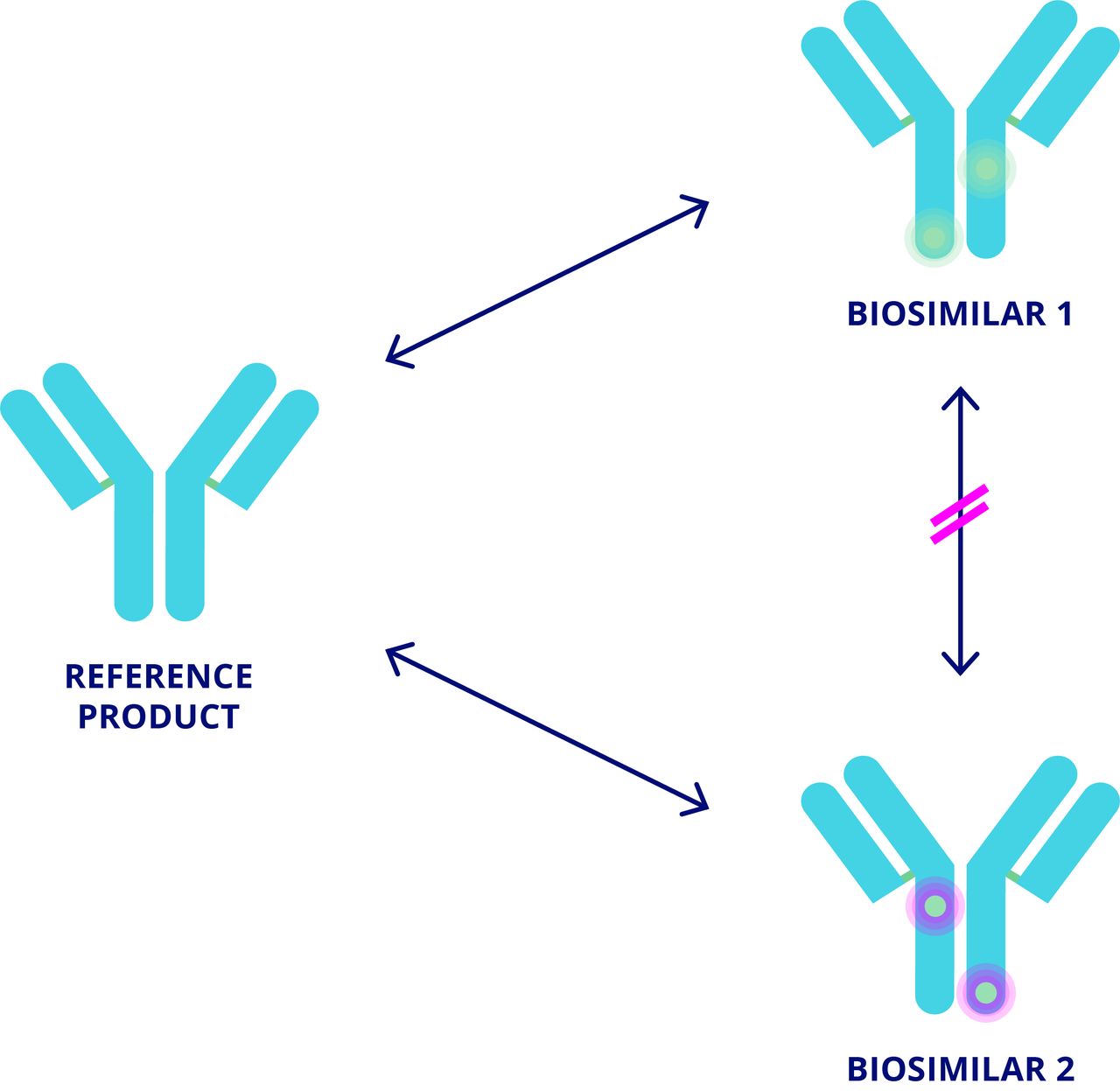
{kind=link}
Switching between several biologics.
Considering PATIENT management
The key concerns when switching to biosimilars are a potential loss of efficacy, changes in immunogenicity with potential secondary failure due to anti-drug antibodies (ADAs), or unanticipated differences in the safety profile compared with the reference product. In addition to unanticipated adverse events, safety considerations include the potential for acute reactions such as injection site reactions (ISRs) or infusion related adverse events compared with the reference product. An awareness of these potential issues by both patients and physicians is of paramount importance. Furthermore, it is imperative that physicians who have jurisdiction over whether to switch from reference to biosimilar have good understanding of important concepts with respect to biosimilarity.
Clinical studies of reference agents versus their biosimilars support maintained efficacy and comparable safety profiles between a biologic and its biosimilars. However, physicians and patients should be aware that biosimilars may be indicated for use in some disease settings without having been evaluated in the relevant target population. For the assumption of comparable efficacy and safety across different pathogenic conditions to hold true, it is necessary to understand how biological therapies exert their effects. For example, tumour necrosis factor (TNF) inhibitors seem to have several distinct mechanisms of action (MOA) that vary by disease setting. MOAs include binding to soluble TNF, membrane bound TNF, and Fc receptors, and it is likely that other unknown MOAs exist.17–21
The key concern when switching to biosimilars is the potential for increased immunogenicity. Clinicians need to be made aware that the immunogenicity of biosimilars can theoretically differ from that of their reference agent (comparative data should be available) or from that of another biosimilar based on the same reference agent (comparative data are not available). It is well established that the persistent presence of an antigen induces an initial immune response followed by tolerance and that conventional B cells and T cells are anergised (ie, functionally incapacitated) when an antigen is constantly present.22 23 Conversely, the intermittent presence of an antigen (or epitope) promotes a persistent immune response that is similar to that observed in vaccination and vaccine recall.23 Therefore, subtle changes in the protein structure or delivery of a biosimilar (eg, dose, dosing regimen, route of administration, or vehicle) can potentially affect the level of immunogenicity observed. ADAs develop in up to a third of patients treated with biologics; neutralising antibodies can reduce an agent’s efficacy, alter their pharmacokinetic profile, and lead to hypersensitivity reactions.23–25
Clinicians also need to be aware of the potential for misattributing secondary failures (ie, due to ADAs) to the substitute agent as the onset of an adverse reaction or secondary failure can be delayed. One preventive measure to avoid this could be the routine screening of patients for the presence of ADAs before switching. This approach is feasible given the availability of highly sensitive assays for ADAs.24 Additionally, if ADAs are detected during treatment with the reference product, switching to a biosimilar may not be appropriate. However, additional tests for ADAs are also associated with increased costs. Manufacturers have a role in providing the necessary clinical diagnostic support to enable the physician and patient to make the correct decisions regarding ongoing therapy.
Despite the possibility that changes in immunogenicity can theoretically lead to a poorer clinical outcome or secondary failure, there is little clinical evidence to support this occurring to date. Enbrel seems to have slightly higher rates of immunogenicity than the biosimilars SB4, GP2015, and CHS0214,26–28 although conclusions cannot currently be drawn because immunogenicity does not seem to affect efficacy or safety in most of the biosimilar trials, and high concentrations of drug may interfere in the assay and lead to false negative ADA results29 (table 1).
Comparable immunogenicity between different etanercept biosimilars
Switching from etanercept to SB4: a case study
A phase III study comparing the similarity (in terms of safety and efficacy) of the reference biologic etanercept (Enbrel) with the biosimilar SB4 (Benepali) in patients with rheumatoid arthritis reported significantly lower rates of ISRs with SB4 versus etanercept at 24 weeks (3.7% v 17.2%, respectively; p<0.001).30 It is suggested that this finding could possibly be due to differences in the formulation; in contrast to etanercept, SB4 does not contain L-arginine and the needle shield does not contain natural latex. Although the finding was supported by the 52 week data,31 the European Medicines Agency concluded that it likely occurred by chance.28
An increased incidence of hepatobiliary disorders was also noted in the European public assessment report for this study, with a greater number of patients experiencing events in the SB4 versus the etanercept arm (11 v 0 patients, respectively).32 However, further investigation revealed that biliary risk factors were also more common in the SB4 arm compared with the etanercept arm, and it was concluded that the observed difference in hepatobiliary adverse events was not related to treatment.28
Subsequently, a long term, open label extension of the same study specifically addressed the feasibility of switching from etanercept to SB4. The results showed no loss of efficacy for up to 100 weeks, with a similar efficacy observed in the switching and maintenance groups (table 2).33 Furthermore, switching did not result in any treatment emergent problems or an increase in adverse events or immunogenicity (table 2).33 Overall, SB4 was well tolerated and effective over 2 years in patients with rheumatoid arthritis.33
American College of Rheumatology response rates, safety, and immunogenicity during the open label extension period of study NCT01895309 that examined switching of etanercept to SB4 versus maintenance of SB4
Existing and future sources of evidence
Data from clinical trials and real world observational studies will increasingly become available to facilitate evidence based clinical decision making in relation to switching (table 3).34–36 For example, data from clinical trials that evaluated switching from adalimumab to the biosimilars ABP501 (psoriasis) and SB5 (rheumatoid arthritis) have recently been published. In both of the studies, overall efficacy, safety, and immunogenicity were comparable in patients who switched from adalimumab to biosimilar and in those who were maintained on adalimumab.37 38 Similarly, a growing body of evidence supports comparable efficacy, safety, and immunogenicity in patients with rheumatoid arthritis whether switched from infliximab to the biosimilar CT-P13 or maintained on infliximab. The evidence base comprises data from randomised clinical trials39 40 and observational real world studies (table 3).41 42 As clinical trials that specifically address the switching between reference agents and biosimilars become more common, increasing efforts should be made to standardise the design and reporting of outcomes for these studies. Despite concerns about switching from a reference product to a biosimilar, reports do not indicate that biosimilar treatment is associated with increased risk.43
Studies of reference and biosimilar infliximab, etanercept, adalimumab, and rituximab incorporating a switch protocol
Considering pharmacovigilance
Post-marketing surveillance of the complex emerging biosimilars market is critical. As biosimilar treatment options expand, clinicians and patients must be better prepared to identify and report new signals. Existing systems for pharmacovigilance, including registries and adverse drug reaction databases, are likely to be insufficient. For example, registries permit the traceability of biologics to the manufacturer, but they do not support the routine capture of information relating to specific batches.44 Equally, the existing ‘yellow card’ systems and adverse drug reaction databases require the time intensive completion of paperwork, which may not be feasible as an additional administrative burden on top of everyday clinical practice.
Improvements to the pharmacovigilance system
All dispensing information (eg, drug (including batch information), dose/frequency/duration, patient identifier) and potential adverse effects associated with biological agents (reference products and biosimilars) should be reported to national authorities by pharmacies and healthcare professionals, respectively. The data should then be collated and analysed on a regular basis to identify potential signals. Importantly, relevant findings need to be fed back to clinicians and patients such that clinical practice is adjusted to address any issues.45 To facilitate data collection, existing systems need to be streamlined and online reporting platforms should be established.
To date, the European experience of biosimilars in other therapeutic fields, in terms of scientific credibility and regulatory decision making, has been good,46 47 but to ensure robustness of safety through the whole of the therapeutic cycle, pharmacovigilance systems must evolve with respect to appropriate naming and traceability.47 Moreover, there will be a need for greater public and professional awareness. To this end, information on pharmacovigilance should be disseminated via medical schools, national societies, medical charities, and governments.45
Considering overall economic benefit
As biosimilar manufacturers target more complex molecules and comply with increasingly stringent regulatory requirements, development costs will likely increase. The cost of biosimilars in relation to the reference product is principally governed by the pricing structure of the regional healthcare system. However, increasing product development costs will inevitably reduce the price differential between reference biologic and biosimilar, thus limiting potential cost savings. The designation of products as interchangeable will also have a significant impact on market penetrance by biosimilars and may be crucial for long term uptake.48
As the differential narrows, it is important that the overall costs of NMS to biosimilars are balanced against the potential risks to the patient. Following approval, biosimilar products may use different administration routes or injector devices compared with the reference product. Consequently, additional patient training may be needed when switching between products, which requires additional time from physicians, nurses, or patients and is associated with cost implications. Additionally, given the need for increased pharmacovigilance, NMS to biosimilars may require additional monitoring or clinical tests, which are also associated with a cost. In the case of intended copies, these costs may be even higher as greater vigilance is required. In summary, these potential hidden costs should be factored into the overall health economic evaluation when considering NMS to a biosimilar.
Considering the future
Patients and clinicians need to understand the potential cost and clinical implications of switching to biosimilars, but almost a decade of experience in clinical practice has thus far raised few concerns.46 Data from clinical trials and real world observational studies that specifically examine switching continue to accrue, providing a growing evidence base to aid clinical decision making. As the availability of biosimilars increases, NMS will serve to reduce overall healthcare costs and permit access to biological agents for a much wider global patient population. In doing so, there is a need for improved pharmacovigilance systems. A greater public and professional awareness of the need and value of pharmacovigilance should be fostered through the dissemination of information to patients, physicians, and payers, as well as provision of education by medical schools, national societies, medical charities, and governments.