Article Text

Abstract
With the authorisation of an increasing number of biosimilars, and the prospect of multiple biosimilar switching, biosimilar naming and the importance of this for pharmacovigilance are coming into sharper focus. Current naming policies are not universal; neither are extrapolation criteria. Indeed, consideration of whether we can extrapolate information from one indication or disease to another continues to be a divisive topic. However, this is changing, as we strive for a more harmonised approach.
Such a unified approach will be needed when considering future strategies to follow for multiple biosimilar switching, especially so because there is currently no uniform policy regarding interchangeability, switching, and automatic substitution. In this multiple biosimilar setting, the question as to whether we can be confident to move across indications will be increasingly important. The cost of biosimilar switching also needs to be considered—biosimilar use may mean that patients need more training and medical visits, with associated administrative costs.
The biosimilars debate seems to be refocusing issues that have previously been extensively discussed but that have recently lost impetus, including the role of clinical pharmacology in internal medicine.
Statistics from Altmetric.com
Considering the biosimilar name
Biosimilar naming to allow biosimilars to be clearly distinguished from their reference product is important to allow physicians to prescribe the intended biological medicine; it is also crucial for reporting adverse effects that may be observed for both the original and biosimilar drugs. Furthermore, giving the intended biological medicine to the patient at the pharmacy level needs to be ensured.
The ability to distinguish a biosimilar from the reference product, or from another biosimilar, has a bearing on accurately tracking the biological medicine.1 If there is clarity on naming, the product can be followed as it is switched from innovator to biosimilar, or even when many switches may be made such as between several biosimilars.2
There is a debate about naming: the first idea is to have distinct, non-proprietary names,3 in which it is acknowledged that the biosimilar is similar but not identical to the innovator. This ensures prescription of the correct medicine and facilitates biosimilar tracking. The second idea is to use identical non-proprietary names for both biosimilar and reference product. Here, identification of the pharmaceutical product is by its active pharmaceutical ingredient.4 There are several possible types of identifier, such as brand and batch number, which can be used to identify and track post-manufacturing changes and to enable determination of whether different biosimilars are being made. However, it is remarkable that we do not have a clear perception about the impact of manufacturing changes on the safety and efficacy of original drugs in order to understand whether different original drugs have been manufactured during their cycle. Therefore, a clear opportunity now exists with the biosimilars to implement an efficient pharmacovigilance system that can assess the impact of biologics manufacturing on the efficacy and safety of drugs.
Considering naming policies
Policies differ between agencies (table 1).5 In Europe, biosimilars are authorised by the European Medicines Agency (EMA) and interchangeability is authorised by individual member states. In Europe, there is a ‘prescription by brand’ policy to distinguish the actual product given to patients and enable pharmacovigilance.3 For example, for etanercept, the innovator is Enbrel and the biosimilar Benepali. However, for both biosimilar and innovator, the same international non-proprietary name (INN)—etanercept—is used.
Nomenclature differs between agencies
In 2015 the US Food and Drug Administration (FDA) finalised a formal naming policy in which a ‘family name’ identifies the biosimilar; however, interchangeability requires additional information, so a four letter suffix strategy is applied.6 For example, Zarxio, the first biosimilar licensed in the US, and the only biosimilar commercialised, was given the name filgrastim-sndz. Other examples are shown in table 1.
Australia is currently reviewing its biosimilar naming policy, but in the interim the INN of the reference product is used.7 This is also the case in Japan, but with the addition of the letters ‘BS’ and a number corresponding to the order in which the biosimilar was approved, to allow non-innovator products to be identified.3
Considering a unified approach to naming
The World Health Organization (WHO) is trying to harmonise the naming of biosimilars to prevent an escalation in the number of different national naming systems.8 As with FDA policy, the WHO proposes a biological qualifier be added to the name of the product, the purpose of which is to complement the INN by adding a unique four letter code that identifies the manufacturer of the active substance. It proposes that this be applied to all biological products—innovators and biosimilars. However, this is not a mandatory policy and so relies on voluntary participation by individual countries; currently, there is no agreement between them.
Appropriate naming is important for patients to feel comfortable with biosimilars. In Europe, many trade associations and patient advocacy groups are calling for more transparency, to have a better way to distinguish whether a patient is being prescribed a biosimilar or innovator.9–11 In the biosimilar prescribing information (package insert), patient information leaflet, and summary of product characteristics (SmPC), only information about the reference product is included.9 10 Biosimilars’ SmPCs do not include information about the clinical trials that were conducted for biosimilar approval.12 13
Considering the clinical equivalence margin
For biosimilar approval, clinical equivalence studies are required. These are to demonstrate that the biosimilar’s efficacy is within a pre-established range of the innovator. However, there is no uniformity with respect to the strategies to determine equivalence for anti-tumour necrosis factor (TNF) agents.
For the infliximab biosimilar CT-P13, when compared with innovator infliximab, the EMA required the 95% confidence intervals for the percentage of patients achieving the American College of Rheumatology (ACR) 20 response to be within an equivalence range of ±15%.14 Similarly, for the etanercept biosimilar SB4, the EMA had a very similar criterion to the FDA—95% confidence intervals for ACR20 within an equivalence range of ±15%.15 However, despite the study for ABP501 being in the same patient population and considering the same response—patients with rheumatoid arthritis treated with methotrexate achieving ACR20—and although it is known that infliximab and adalimumab have similar clinical performances, the FDA recommended a different strategy for equivalence. That is, the 90% (not 95%) confidence intervals should be within an equivalence range of ±12%.16 Finally, for the etanercept biosimilar GP2015, the pivotal clinical study was performed in patients with psoriasis treated with monotherapy. The outcome was Psoriasis Area and Severity Index (PASI) 75 (75% improvement from baseline PASI score), and the FDA recommended that 95% confidence intervals should be within an equivalence range of ±18%.17
The consequences for indication extrapolation of having different equivalence ranges for anti-TNF agents is currently unknown.
Considering indication extrapolation
Extrapolation—to extend information and conclusions available from studies of medicinal products in one or more subgroups of the patient population (‘source population’) to make inferences for another subgroup of the population (‘target population’), or condition or medicinal product—reduces the need to generate additional information.18
For biosimilars, a complete dossier of Chemistry, Manufacturing and Control information exists, containing extensive information about the quality, characteristics of the products, pharmacological data in all mechanisms of action, and information regarding clinical studies in a specific indication. From this indication, it may be possible to extrapolate to other indications/diseases.19 However, to be able to do this, clinical studies should be performed in a sensitive population.
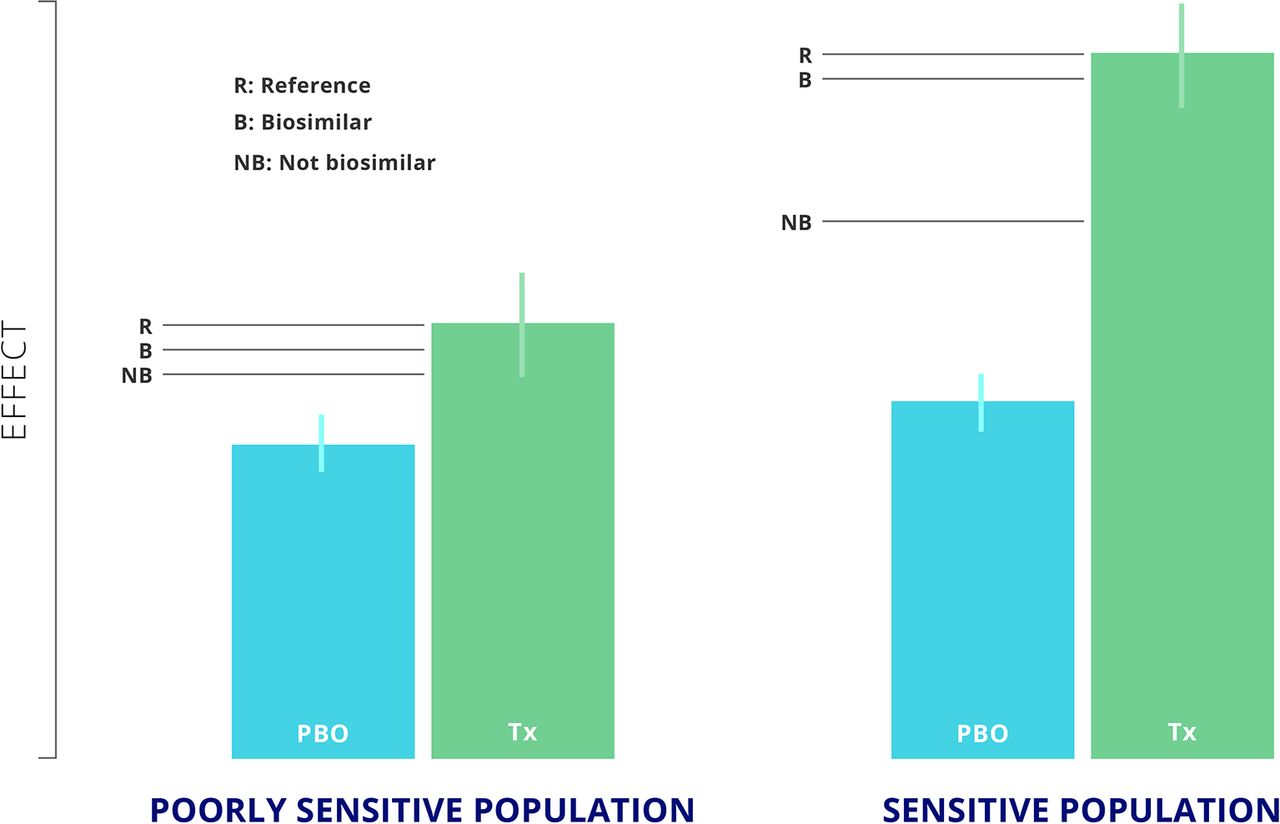
A sensitive patient population is one in which there is a clear response with respect to placebo or control conditions. The reference product can be distinguished from non-biosimilar product, but the biosimilar will yield a very similar or equivalent response to that of the reference product (figure 1).20 In contrast, in a poorly sensitive population, although the reference product, biosimilar, and a non-biosimilar product may each yield a significant difference with respect to placebo, it would not be possible to differentiate the non-biosimilar response from that of the biosimilar and reference products (figure 1).20

The difference between a sensitive and poorly sensitive population adapted from Castañeda-Hernández, 2015.
We can illustrate this using the example of trastuzumab in the treatment of breast cancer. Jackish et al suggest that the neo-adjuvant/adjuvant population is a homogeneous and sensitive population to establish similarity for a trastuzumab biosimilar candidate.21 However, a different approach has been taken by Mylan, a biopharmaceutical company that performed a clinical study in the metastatic population and submitted the results to the FDA.22 It remains to be seen whether the FDA will accept the results from this more heterogenous population.
Issues associated with extrapolation
In general, it is accepted that extrapolating from the oncology to the inflammatory diseases setting is not valid. Thus, different clinical assessments need to be carried out for drugs such as rituximab in patients with cancer, with a different set of data required for patients with inflammatory diseases (table 2).23
EMA and FDA response to concerns regarding extrapolation of clinical data
For anti-TNF agents in inflammatory diseases, it is not clear which is the most sensitive indication from rheumatoid arthritis, Crohn’s disease, and psoriasis. Moreover, for patients with rheumatoid arthritis, it is also not clear which are the most sensitive populations, whether methotrexate-naïve patients, patients with an insufficient response to methotrexate, or those with insufficient response to other biological agents.
There are no uniform criteria for extrapolation among different regulatory agencies. For example, in South Korea, authorisation for CT-P13 was granted in rheumatoid arthritis and ankylosing spondylitis with a complete set of clinical trials, and authorisation was granted for psoriasis, psoriatic arthritis, Crohn’s disease, and ulcerative colitis by extrapolation only.14 24 25 A similar policy was followed by the European Union and the FDA.26 27 However, Health Canada had a different view, with additional data being required to authorise the product in inflammatory bowel disease (IBD).28
Some of the authors of this article consider that extrapolation should be approached cautiously. Most clinical equivalence studies have been performed in rheumatological indications. However, the pivotal clinical study for GP2015 was performed in patients with psoriasis, and there is concern about using psoriasis (rather than psoriatic arthritis) to extrapolate use of GP2015 to rheumatoid arthritis.
However, there has been substantial uptake of CT-P13 in IBD, and this could be considered an example of clinical practice confirming that indication extrapolation works. However, it should be noted that CT-P13 was tested in ankylosing spondylitis, a disease in which many patients have IBD, and we would be more reluctant to prescribe for rheumatoid arthritis a drug that has been tested in psoriasis but not in rheumatoid arthritis. Therefore, there is an argument for considering extrapolation on a case-by-case basis.
We acknowledge that when discussing extrapolation, there is a need to consider not only what the drug does to the disease (and the potential influence of immunogenicity on pharmacokinetics) but also what the disease does to the drug. Drug responsiveness may vary between diseases, even though drug mechanism may remain the same. Thus, the biosimilars debate seems to be refocusing issues that we should have been considering for many years, including the need to respect the prominence of clinical pharmacology in internal medicine.
Therefore, we have two options: to perform clinical studies for each discipline or to ask the regulatory agencies to communicate not only with guidelines but also by explaining their decisions in terms of drug pharmacology to allow the clinician to make an informed decision. It would be helpful if representatives of the different specialties involved in using biologics and biosimilars could come together to further consider these questions and to exchange experiences and concerns.
Considering interchangeability, Switching, and automatic substitution
Interchangeability, switching, and automatic substitution, although similar concepts, are not identical.
Interchangeability
Interchangeability is the practice of alternating between one medicine and another without significant risk of an adverse health outcome.1 29
According to the FDA, “an interchangeable biological product, in addition to meeting the biosimilarity standard, is expected to produce the same clinical result as the reference product in any given patient, and for a product that is given to a patient more than once, the risk in terms of safety and effectiveness of alternating or switching between the interchangeable and the reference product is not greater than the risk of using the reference product without alternating or switching.”30 Data are now required on three switches between products to establish interchangeability.31
Differences exist between regulatory guidelines used in different countries (figure 2).30 32–35 There are currently no official guidelines for interchangeability studies.1

Interchangeability designation – heterogeneity of regulatory guidelines.
Switching
Medical switching is a physician’s decision to exchange one medication for another, usually to optimise treatment or to minimise adverse events.29 36 37 The substitute agent is typically, though not always, introduced after the first agent has failed, with patients switched to a substitute agent usually remaining on that agent.
In contrast, non-medical switching, in which a substitute agent is typically introduced in stable patients, is driven by economic reasons and may be enforced through guidelines and/or legislation. Therefore, a prescribing physician may not be a part of the non-medical switching decision.37 Efficacy, safety, or convenience benefits are not expected.
Automatic substitution
Finally, automatic substitution is when a pharmacist is authorised to substitute a prescribed medicine without the consent of the prescribing physician.1 29 As for interchangeability, there are differences between regulatory agencies (figure 3).1 38 39 In Europe, most member states did not allow automatic substitution of biological medicines,1 but this is changing quite rapidly.40 41

{kind=link}
{kind=link}
{kind=link}
Automatic substitution—worldwide policies.
Data considerations
Norway recently completed a large interchangeability study, NOR-SWITCH, in which inflximab was interchanged for the biosimilar infliximab CT-P13. Patients stable on infliximab in all authorised indications—rheumatology, dermatology, and gastroenterology—were randomised to either continue with the innovator infliximab or to switch to CT-P13. The findings showed that, in terms of infliximab trough concentrations, there was no significant difference between innovator and biosimilar.40 Also, concerning the presence of anti-drug antibodies, the results were similar between innovator and biosimilar.40 For the overall population, the results were within the pre-established equivalence margin (±15%), but because patient numbers were low for the individual indications, it was not possible to draw conclusions for these populations.42
Three month real life data from the DANBIO study of 693 patients with rheumatoid arthritis, psoriatic arthritis, or ankylosing spondylitis have also been reported.41 Here, patients were switched from infliximab to CT-P13 by mandatory non-medical switching. Overall, there was no significant difference between innovator and biosimilar after switching. However, 45 (7%) patients stopped CT-P13 during their 3 months’ follow-up, and 20 patients lost efficacy. This further highlights the need for stringent pharmacovigilance after switching.41
For extrapolated indications, the clinical data that we have currently are not consistent with respect to switching. For example, although extrapolation from rheumatoid arthritis to IBD has been supported by clinical data,43 44 some studies have not yielded satisfactory results. For example, in a prospective, multicentre nationwide cohort study performed in Hungary, patients switched to CT-P13 had decreased response rates and were more likely to develop allergic reactions than patients naïve to infliximab.45
Cost considerations
In addition to more medical visits, increased administrative costs, potential supply failures, and emergency acquisitions, patients may need to be trained to use a different device. However, with respect to injection device and patients’ satisfaction, switching does not seem to be a source of concern. In a recently published paper, in which nurses were grading patients’ satisfaction with the Benepali biosimilar autoinjector, the latter was rated better than the innovator’s autoinjector.46
Considering the future for biosimilar policy—a summary
Clear naming strategies and traceability are needed for biosimilars. It is important to follow when the switch was performed and how it was performed. The prospect of multiple biosimilar switching further highlights the need for biosimilar naming policies that facilitate pharmacovigilance.
Extrapolation criteria are not universal. However, these are evolving towards harmonisation, and we hope that this will soon be achieved. In addition, criteria for interchangeability and automatic substitution are currently not universal.
The question of whether we can confidently move across indications or diseases will become increasingly important when we enter the world of multi-biosimilars and multiple biosimilar switching. For example, in just a few months switching may occur between innovator etanercept and two or more biosimilars, as two biosimilars are already authorised by the main regulatory agencies. We do not yet have any clear criteria for multiple biosimilar switching. So, what strategy should we follow, especially when dealing with extrapolated indications?
We should not be afraid of biosimilars, although the same is not the case for intended copies. By relating data clearly to clinicians, manufacturers and regulators will be possible to offer them if not certainty then at least confidence in the use of biosimilars.