Article Text

Abstract
As of mid-2017, 10 tumour necrosis factor inhibitors (four for etanercept and three each for adalimumab and infliximab) and a first rituximab biosimilar are on the market, and a considerable number more are in various stages of development. The clinical trials of biosimilars, which have included long term extensions, have used various designs to look at switching between originator and biosimilar products, with reassuring results for clinical practice. For the infliximab biosimilar CT-P13 in particular, several studies have examined non-medical switching in real world practice. The results suggest that switching does not compromise safety, efficacy, or immunogenicity. However, additional data from clinical and real world switching studies, especially of switching between two or more biosimilars, are needed, as is continuing pharmacovigilance with larger databases to fill remaining gaps in the evidence. As biosimilar development continues, innovations in formulation and drug delivery technology may become of increasing interest.
Statistics from Altmetric.com
Considering the current status of biosimilars
A survey conducted in 2015 found many potential tumour necrosis factor (TNF) inhibitor biosimilar agents at various stages of development, especially for etanercept and adalimumab (eight and six respectively), but also four for infliximab and the first for tocilizumab.1 The pipeline for biosimilars is still increasing.2 In the 2015 survey, the number of rituximab biosimilars being developed was even larger at 10, reflecting the speed of development of biosimilars both within and beyond rheumatology. Up to 2015, data had been published on seven biosimilars for adalimumab, four each for etanercept and infliximab, and three for rituximab.3 Figure 1 4 5 shows that there are additional biosimilars undergoing development.
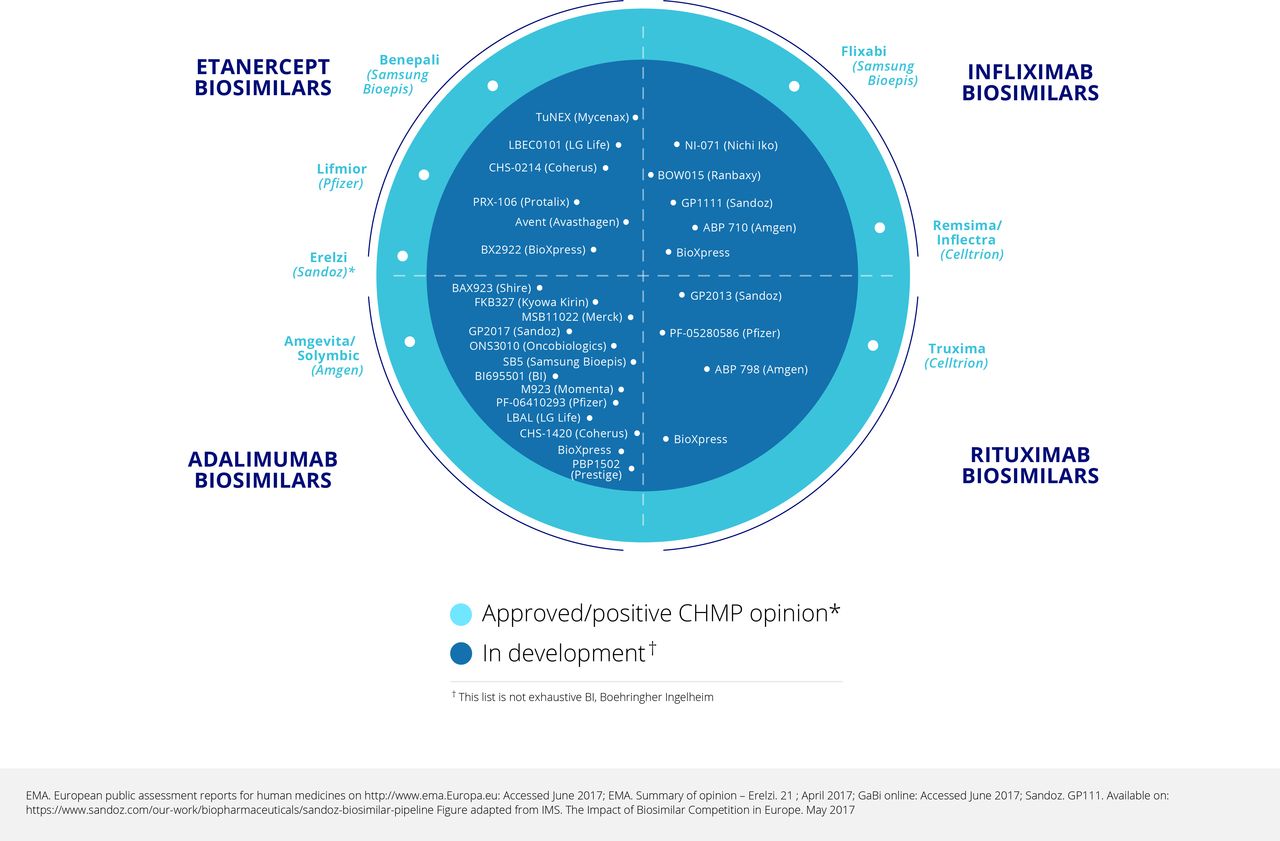
Biosimilars approved, and in development: May 2017.
It is necessary to distinguish true biosimilars from the so called ‘intended copies’ or ‘biomimics’ that are on the market in some parts of the world, often at a very low price and with uncertainty about efficacy. Two such products for rituximab and six for etanercept were found in a survey conducted in 2015.1 It is important to note that these products have not been developed according to biosimilar regulatory guidelines.
The goal of the pre-clinical studies is to match the biosimilar candidate to the reference product as closely as possible through an extensive ‘comparability exercise’ from analytical characterisation of critical quality attributes and testing through clinical studies.6 These human studies then provide clinical confirmation of equivalent safety and efficacy, which should address residual uncertainty left by preclinical comparability exercises. As required by the regulators, these studies are not designed to demonstrate superiority or inferiority, which is not the intention of a biosimilar development.6 As of mid-2017, 10 TNF inhibitor biosimilars and one rituximab biosimilar had been approved in various markets (table 1).7
A snapshot of licensed biosimilars: January 2017
Considering clinical data for approved biosimilars
Etanercept biosimilar: SB4
Following the preclinical comparability exercise, the pharmacokinetics, safety, and immunogenicity of SB4 were compared with US and EU Enbrel in 46 healthy volunteers.8 This double blind, randomised, three part, two period, two sequence, single dose, crossover study used healthy volunteers to reduce heterogeneity and maximise the opportunity to detect any differences.
This study was followed by a clinical trial in patients with rheumatoid arthritis (RA), comparing SB4 and EU Enbrel 50 mg once weekly on a background of methotrexate. This was a typical example of a biosimilar study, with a 52 week randomised double blind period followed by an open label extension period up to week 100 in which all patients received SB4 and methotrexate allowing evaluation of a single switch from a bio-originator to the biosimilar.9 The double blind period included 596 patients, but the extension period was carried out only in Poland and the Czech Republic and included 245 patients.10 The primary endpoint was American College of Rheumatology 20 (ACR20) response rate at week 24. This was achieved by 78.1% of patients on SB4% and 80.3% on Enbrel in the per protocol set—a difference of –2.22 (95% CI –9.41 to 4.98), which is within the predefined equivalence margin of –15% to 15%.11 The difference in the full analysis set was 1.92 (−5.24 to 9.07). The ACR20, 50, and 70 response curves up to week 52 were almost superimposable for the two products (figure 2).9 Radiographic damage, not an absolute requirement of biosimilar studies, was also assessed; the proportion of patients with change in modified Total Sharp Score (mTSS) at week 52 greater than the smallest detectable change (2.3) was lower with SB4 than Enbrel (8.4% v 14.0%; p=0.050).9 However, this was mainly driven by joint space narrowing and is still considered to demonstrate equivalence.

ACR response rates for SB4 and etanercept up to week 52. ACR, American College of Rheumatology; ETN, etanercept; FAS, full analysis set.
The overall safety profiles of SB4 and Enbrel were comparable, with 58.5% and 60.3% of patients respectively reporting at least one treatment emergent adverse event (TEAE). Serious infections were reported in 0.3% and 1.7%, respectively, and malignancies in 1.3% and 0.3%.9 One interesting difference was in injection site reactions, which were reported in 3.7% of patients taking SB4% and 17.5% of those taking Enbrel. Another interesting finding is that hepatobiliary adverse events were reported in 11 (17%) patients in the SB4 arm but none in the Enbrel arm.8 12 However, examination of individual data showed that more patients in the SB4 groups had biliary risk factors; these were not defined further. It was concluded that the observed difference in the incidence of hepatobiliary events was not treatment related, and the European Medicines Agency (EMA), US Food and Drug Administration (FDA), and other agencies have approved SB4.
The other area that has given rise to discussion is the incidence of antidrug antibodies (ADAs) in the SB4 study. The cumulative data show an incidence of 0.7% for SB4% and 13.1% for Enbrel.11 However, this reflects a transient increase in ADAs in the Enbrel arm at weeks 4 and 8, followed by a return to zero from week 16. The nature of this transient increase in ADAs is not clear. Notably, these antibodies were not neutralising; subgroup analysis showed no relation between ADAs and injection site reactions, and the overall efficacy and safety profiles of the two products are similar.
Etanercept biosimilar: GP2015
As for SB4, the pharmacokinetics, safety, and tolerability of GP2015 were first studied in healthy volunteers.13 14 Results from a clinical trial in patients with psoriasis (EGALITY) were presented in 2016,15 and a further trial is ongoing examining the efficacy, safety, pharmacokinetics, and immunogenicity of GP2015 compared with Enbrel in patients with RA.
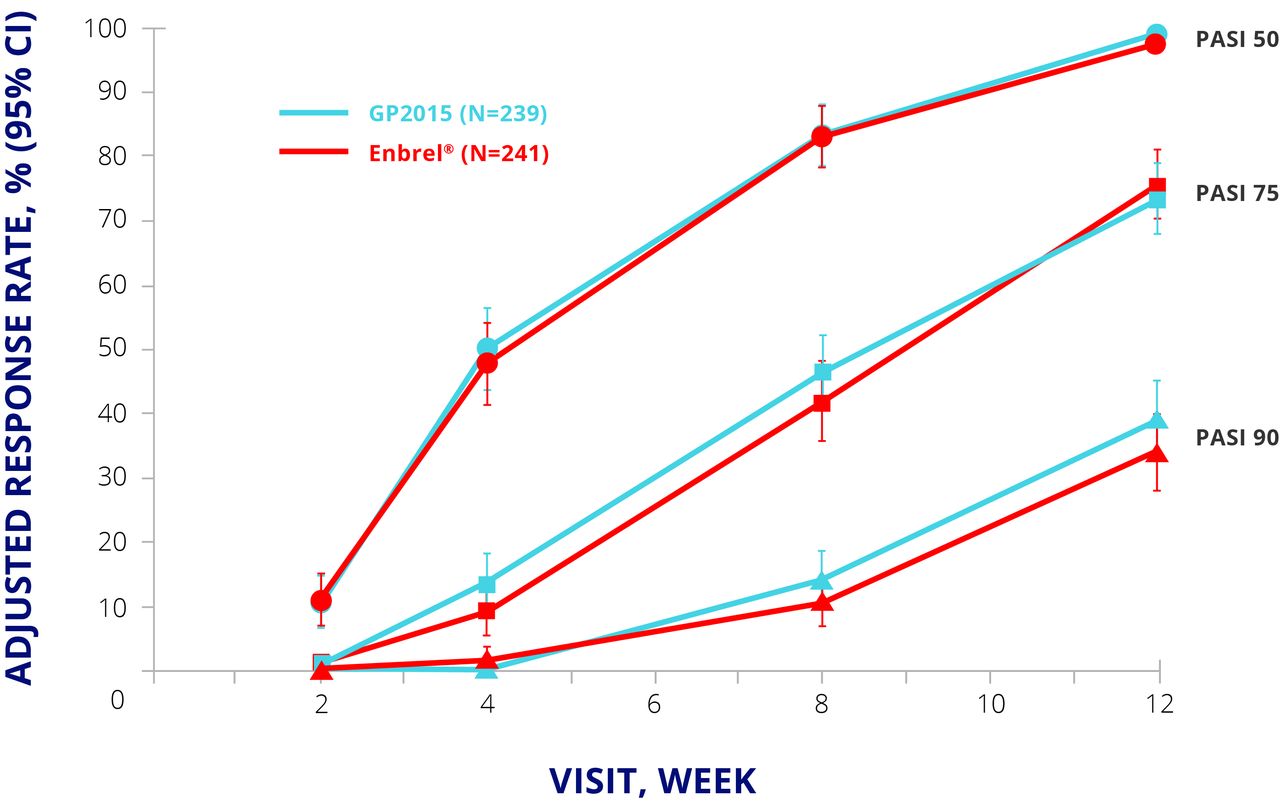
EGALITY randomised 531 patients with moderate to severe psoriasis to GP2015 or Enbrel 50 mg twice weekly for an initial 12 weeks and 50 mg once weekly thereafter. Patients who achieved a Psoriasis Area and Severity Index (PASI) 50 response at week 12 were eligible to enter period 2, in which they either continued their allocated treatment or were switched to the other drug. The switching arm involved two further switches at weeks 18 and 24; from week 30, patients could continue their current treatment in an extension phase up to week 52. The primary endpoint was PASI 75 response at week 12.15 The primary endpoint was met, and PASI 50, 75, and 90 responses were similar up to week 12 (figure 3).15 The safety profiles for both products were similar to published data for Enbrel, with no major differences in TEAEs of special interest such as infections and neoplasms.15
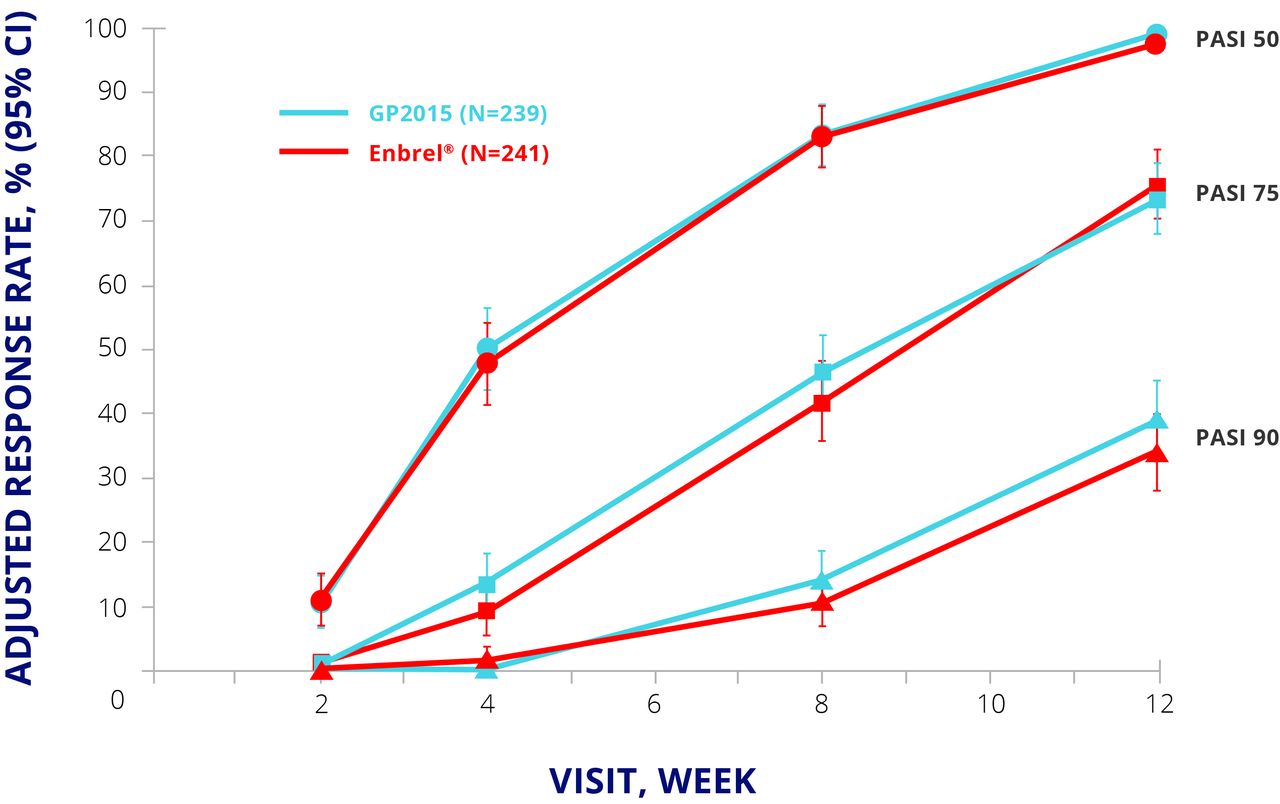
EGALITY: PASI 50, 75, and 90 response rates at week 12. CI, CI interval; ETN, etanercept; PASI, Psoriasis Area and Severity Index; PPS, per protocol set.
Notably, RA and psoriasis have been considered as ‘sensitive populations’ (regulatory term) in such phase III trials for etanercept biosimilars, to detect differences compared with Enbrel in these phase III programmes.
Immunogenicity
As in the SB4 RA study, GP2015 showed lower immunogenicity than Enbrel, with ADAs detected in five (1.9%) Enbrel patients at week 8 compared with none for GP2015. Interestingly, similar data have been seen with CHS0214, another etanercept biosimilar, for which the week 24 assessment showed an ADA rate of 1.3% compared with 4.7% for Enbrel.16 The overall tendency seems to be that Enbrel has slightly higher rates of immunogenicity than the biosimilars, although firm conclusions cannot currently be drawn. In addition, secondary failure of etanercept (unlike the monoclonal antibody TNF inhibitors) does not depend on ADA formation.
The assays used to detect ADAs are becoming increasingly sensitive and more specific, and immunogenicity does not seem to affect efficacy or safety in most of the biosimilar trials. We highlight the importance of stating the assay methods when reporting immunogenicity results. It is also important to measure and report drug trough concentrations and to examine the pharmacokinetics and immunogenicity together. For example, in the SB4 RA clinical trial, trough concentrations of SB4 were higher than those of Enbrel; these high concentrations may have interfered in the assay and led to false negative ADA results. Assays that include acid dissociation enable more accurate ADA determination in the presence of high drug concentrations. We believe that drug and ADA concentrations should be measured every six months as part of pharmacovigilance in the registries. However, although measuring ADA is necessary for active pharmacovigilance, we suggest that it is not needed for a clinical decision.
Infliximab biosimilar: SB2
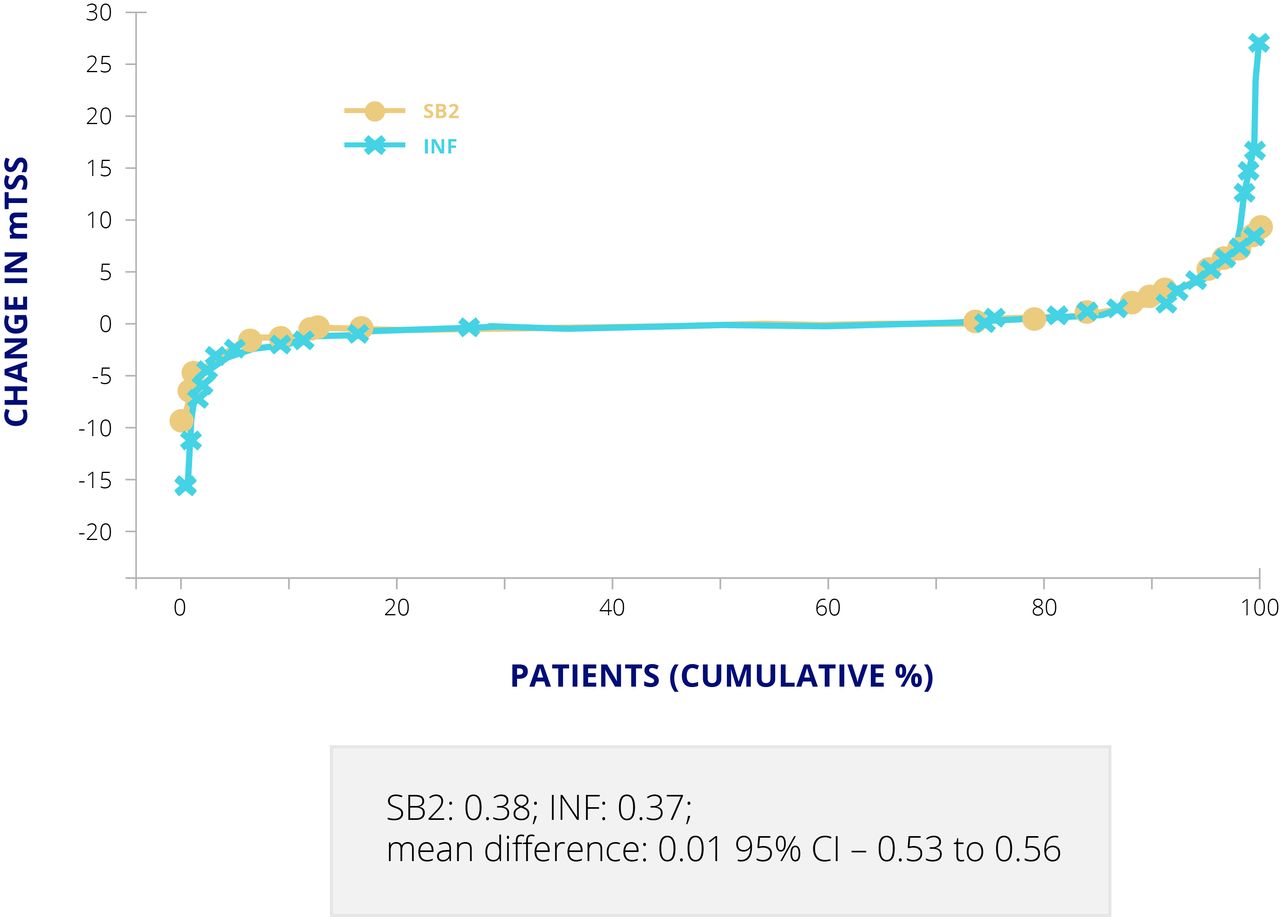
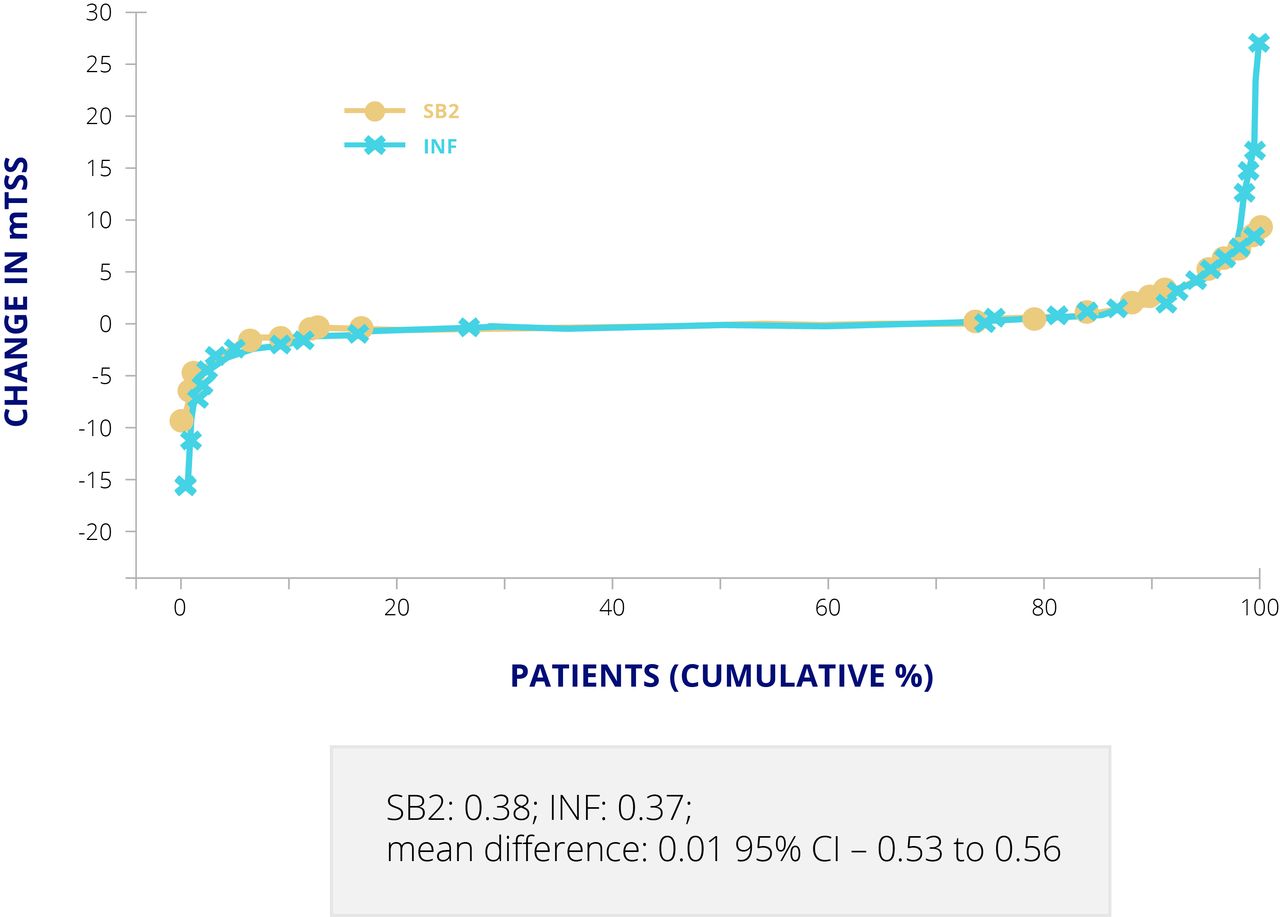
For the infliximab biosimilar, SB2, approved by the EMA and FDA, a double blind, randomised, parallel group study was carried out to compare SB2 and Remicade, both at 3 mg/kg on a background of methotrexate, over 54 weeks in 584 patients with moderate to severe RA.17 ACR20 response at week 30, the primary endpoint, was almost identical in the two groups, at 64.1% for SB2% and 66.0% for Remicade (adjusted difference −1.88, 95% CI −10.26 to 6.51) in the per protocol set (55.5% v 59.0% (adjusted difference −2.95, –10.88 to 4.97) in the full analysis set).17 Cumulative changes in mTSS at week 54 were also comparable (mean difference 0.01, –0.53 to 0.56) (figure 4).17 ADAs at week 30 were detected in 55.1% of patients taking SB2% and 49.7% of those taking Remicade,18 which is typical for infliximab originator and biosimilars. At least one TEAE was reported in 61.7% of the SB2 group and in 65.2% of the Remicade group.18
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cumulative changes in mTSS for SB2 and infliximab. Values are presented as mean (SD) in patients with available mTSS at both baseline and week 54. INF, infliximab; mTSS, modified Total Sharp Score.
Considering response levels
The ACR response rates in biosimilar trials in RA and the PASI response rates in psoriasis are very high compared with those seen in the pivotal trials for the originator biologics. We suggest more than one explanation for this. One possibility is that many of the participants in the biosimilar studies are from countries in which the healthcare system is not very developed, so a larger response is seen than in patients in more developed countries who have had better previous therapy. Another possibility and a more likely explanation is that the trials do not have a placebo arm, so patients know that they are receiving an active treatment. These enhanced responses have also been noted in bio-originator studies when different active treatments were compared.
Considering extension studies and switching
Biosimilar extension studies and switching studies (exchanging bio-originator for biosimilar) are important for clinical practice. Different study designs have been used to look at switching for the various biosimilars (table 2).19 Some studies have included switching in the double-blind phase, whereas others have conducted open label switches; some studies (such as EGALITY) have a used a crossover design. Switching studies for the infliximab biosimilar CT-P13 include the open label BIO SWITCH study in 200 patients and the double blind NOR-SWITCH study in 500 patients.
Study designs for biosimilar switching: June 2019
For the etanercept biosimilar SB4, the RA clinical trial included a 48 week extension period to evaluate the long term safety, immunogenicity, and efficacy of continuing SB4 versus switching. Of 245 patients from the Czech Republic and Poland enrolled in the extension period at week 52, 126 continued on SB4 and 119 switched from Enbrel to SB4.8 Of these, 119 patients in the maintenance group and 113 switched patients continued to week 100. Efficacy and radiographic progression were sustained and comparable between groups, with response rates of 77.9% in maintained patients and 79.1% in switched patients for ACR20; 59.8% and 60.9%, respectively, for ACR50; and 42.6% and 41.7%, respectively for ACR70.10 The safety profiles between weeks 52 and 100 were also similar, with at least one TEAE reported in 47.8% of patients in the SB4 arm and 48.7% of those in the Enbrel arm.
In the extension of the PLANETRA study of infliximab biosimilar CT-P13, the ACR20, 50, and 70 responses were comparable at weeks 54, 78, and 102 in patients maintained on CT-P13 and those switched from Remicade.20 ADA status and safety profiles were also comparable at week 100, with ADA rates of 46.4% in maintained patients and 49.6% in switched patients, and at least one TEAE in 53.5% and 53.8% respectively.
The SB2 infliximab biosimilar equivalence and transition study also included an extension period in which patients either remained on SB2 or Remicade or were switched from Remicade to SB2 at week 54.17 18 ACR20, 50, and 70 rates were comparable up to week 78 in these three groups, and no differences in safety or immunogenicity profiles were apparent.21
Considering data supporting interchangeability
Interchangeability is a regulatory term, meaning that patients can be changed from one product to another without significant risk of an adverse health outcome.4 22 The FDA issued a guidance document on this in January 2017; data are now required on three switches between products to establish interchangeability.22 The EMA allows individual member states to decide what data are needed to demonstrate interchangeability of biosimilars.
In Norway, a 52 week switch study (NOR-SWITCH) has been conducted in which 481 adult patients on stable treatment with Remicade for at least six months were randomised to continue the same treatment or switch to CT-P13 at the same dose. The primary endpoint was maintenance of clinical efficacy. Disease worsening occurred in 26.2% of patients who remained on Remicade, and 29.6% of those who switched to CT-P13, which was within the pre-specified non-inferiority margin.23 The incidence of ADAs was also similar at 7.1% and 7.9%, respectively. The percentage with disease worsening did not differ between treatment arms in patients with RA, psoriatic arthritis, spondyloarthropathy, psoriasis, or ulcerative colitis. Worsening occurred in 21.2% of Crohn’s disease patients who continued Remicade compared with 36.5% of those who switched to CT-P13.24 However, gastroenterologists do not seem to be concerned about this because of positive worldwide experience with CT-P13 in Crohn’s disease. In a managed switching programme from originator infliximab to biosimilar CT-P13 in 143 patients with inflammatory bowel disease (IBD) (118 Crohn’s disease, 23 ulcerative colitis, and two IBD unclassified), similar patient reported outcomes, biochemical response, drug persistence, and AE profiles were observed before and after the switch.25 Drug acquisition costs decreased by £40 000 to £60 000 per month).
Non-medical switching, whereby a substitute agent is typically introduced in stable patients (driven by economic reasons) was studied retrospectively in the DANBIO registry in Denmark with patients switching from Remicade to CT-P13. Patients with RA, psoriatic arthritis, or axial spondyloarthropathy who switched before 1 February 2016 and had available data on treatment outcomes were included in the assessment (n=768). Mean follow-up time after switching was 336 days.26 Disease activity (DAS28) and disability (Health Assessment Questionnaire (HAQ) score) were largely unaffected by switching. However, 117 (15%) patients discontinued the biosimilar, with adverse events and lack of efficacy cited as the main reasons.
Another observational study of 191 infliximab treated patients in the Netherlands who switched to biosimilar infliximab found that DAS28 remained stable for six months after the switch in RA and psoriatic arthritis patients.27 However, 23% of patients discontinued biosimilar infliximab due to inefficacy (n=35), adverse events (n=23), or infusion reactions (n=2). ADAs were detected in 47% of patients at baseline and in 38% at six months.
Considering biosimilars in development
As of mid-2017, the largest number of biosimilars in development was for adalimumab (figure 1).4 5 There were also six for etanercept (four in clinical and two in preclinical development), five for infliximab (all in clinical development), and eight for rituximab (seven in clinical and one in preclinical development). There is one biosimilar in preclinical development for golimumab, one in clinical and one in preclinical development for abatacept, and three in preclinical development for tocilizumab.7
Developments in biosimilars may increasingly involve improvements in formulation and drug delivery technology. This is already happening—for example, the manufacturer of SB4 removed sodium citrate with the aim of reducing pain on injection; this involved much testing and considerable data to show that this was not associated with increased aggregation. This example illustrates the evolving field of biosimilar development and suggests that although the aim is biological similarity, there is also a drive to innovation.
Considering biosimilars already approved and in development – A SUMMARY
To date, it is reassuring to observe that studies of switches between reference biologics and biosimilars suggest that switching does not compromise safety, efficacy, or immunogenicity. However, additional data from clinical and real world switching studies, especially switching between two or more biosimilars, are needed, as is continuing pharmacovigilance with larger databases to fill remaining gaps in the evidence. National and international registries will play an important role in assessing the safety aspects of biosimilars in the future, and this may be especially important when using biosimilars for indications that have not been examined in clinical trials. For rare diseases for which it may be difficult to recruit patients, future approval may depend only on thorough analytical evaluation of molecules and pharmacokinetic studies.