Article Text

Abstract
Biosimilars are biotherapeutic products with similar efficacy, safety, and quality to a licensed bio-originator. Biosimilars include monoclonal antibodies, soluble receptors, growth factors, and hormones. The manufacture of biosimilars is a sophisticated multi-step process; factors at each stage, such as production cell line, culture conditions, and formulation, may each alter the final product through post-translational modifications. A vial of a therapeutic antibody contains multiple species with distinct glycosylation profiles (microheterogeneity), which are responsible, for example, for complement activation, pharmacokinetics, and structural stability. Whereas the focus for the manufacturer of the bio-originator is to show safety and efficacy in clinical trials, biosimilar development focuses predominantly on in-depth analyses to confirm that the product is identical to the originator in terms of structure, composition, and in vitro activity. Therefore, the critical quality attributes of a biological drug that influences clinical safety and efficacy should be carefully assessed. At least one clinical study is required to compare pharmacokinetics of bio-originator and biosimilar, and at least one sufficiently large randomised controlled trial to demonstrate clinical equivalence. Once biosimilarity is confirmed, regulators may allow extrapolation to other licensed bio-originator indications, provided efficacy relies on a similar mechanism of action in each one. Consequently, a biosimilar may be approved in all indications for which the bio-originator has been approved, without multiple clinical trials.
Statistics from Altmetric.com
Considering biotherapeutics
To understand biosimilars first requires an understanding of biological therapeutics. These treatments, which include growth factors, hormones, soluble receptors, and monoclonal antibodies, are produced in living cells—usually mammalian but sometimes bacterial and even plant cells. This article focuses primarily on monoclonal antibodies.
Antibodies are complex, flexible adapter proteins that link specificity in a so-called variable region to functions such as complement activation, Fcγ receptor binding, neonatal Fc receptor binding, and half life determination in a constant region. Figure 1 schematically represents the structure of an antibody.

Basic antibody structure-function.
The ‘backbone,’ or fundamental structure, of a monoclonal antibody consists of amino acids, which are encoded by DNA and form primary, secondary, tertiary, and quaternary structures, becoming a protein molecule. The cell that synthesises the protein also adds ‘post-translational modifications’ through glycosylation of amino acid residues. Further modifications by methylation, deamidation, and oxidation are introduced during the production and purification process. These modifications are influenced by the cell line chosen to produce the antibody but also by aspects of the manufacturing process and formulation of the drug. For example, the cell culture medium and processes used to purify the antibody from it can both influence important characteristics of the final product. Thus, distinct glycosylation variants of a single monoclonal antibody may differ in complement activation, Fcγ receptor binding, pharmacokinetics (via binding affinity for the neonatal Fc receptor), and structural stability (table 1).1 A vial of therapeutic antibody actually comprises multiple species with distinct glycosylation profiles (microheterogeneity), all products of the cell line used to make the antibody.
Glycosylation may also alter the immunogenicity of a monoclonal antibody and, since glycosylation can influence so many characteristics, regulatory agencies mandate strict control over a product’s glycosylation profile. The same is true for other modifications, but, at present, most is known about the influences of glycosylation. These characteristics of the drug that are known to influence clinical safety and efficacy are called critical quality attributes and are the major focus of drug characterisation.
Examples of modifiable biological critical quality attributes (CQA) of a monoclonal antibody
The manufacture of a novel monoclonal antibody is a sophisticated process, each step of which can influence the final product (figure 2).2 The process begins with identification and cloning of the gene encoding the desired variable region—for example, by using a technique such as phage display. This is then cloned into an ‘expression vector’ adjacent to another gene encoding the selected constant region and subsequently introduced into the selected host cell line. This ‘expression system’ is grown into a ‘master cell bank,’ which is stored securely because of its central and unique importance to the production process. Cells derived from the master cell bank become a ‘working cell bank,’ which will ultimately produce the monoclonal antibody destined to become a therapeutic product. The host cell line, culture medium, and culture conditions are specifically selected to maximise yield of the desired therapeutic product, which is secreted by the cells into the culture medium. After a period of growth, monoclonal antibody is purified from the culture medium and then formulated, packaged, sterilised, and added to vials.
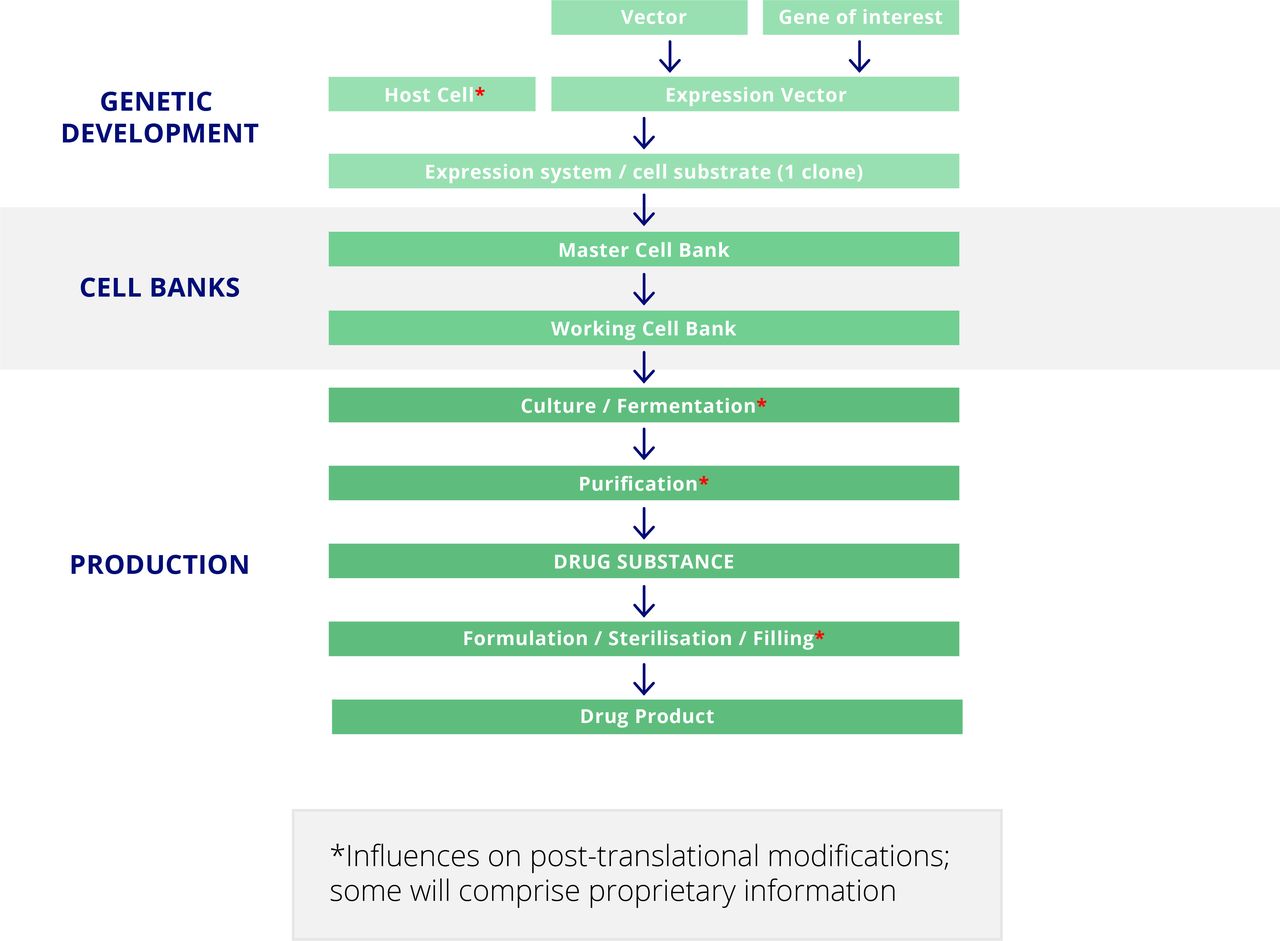
Development and manufacture of a novel monoclonal antibody.
Considering the manufacture and approval of a biosimilar
A biosimilar is defined as a biotherapeutic product that is similar to (has an absence of relevant differences from) an already licensed reference biotherapeutic (bio-originator) in terms of efficacy, quality, safety, purity, and potency.3–5
Small molecule drugs such as non-steroidal anti-inflammatory drugs and corticosteroids are synthesised chemically. Since these drugs have a fixed molecular formula, chemists are easily able to copy them and confirm the identity of the copy to the original version. A manufacturer is granted a license for such a ‘generic’ once it is shown to have an identical structure in vitro (eg, through mass spectrometry) and identical in vivo pharmacokinetics (eg, maximal concentration and half life) to the originator.
A monoclonal antibody is vastly more complex than such a small molecule drug.6 Although the former’s lengthy primary amino acid sequence will be well characterised, post-translational modifications that affect the final product are less predictable because of the aforementioned influences of the expression system and the inherent microheterogeneity. Some of the critical determinants of post-translational modifications may comprise proprietary information and, therefore, not be readily available to biosimilar manufacturers, which therefore face various key challenges. First, they must carefully analyse the bio-originator to understand, as far as possible, its post-translational modifications and its composition. This may vary from batch to batch, particularly when there is more than one bio-originator manufacturing plant—for example, in Europe and the USA. They must then develop a process that consistently results in the manufacture of a product that appears as similar as possible to the bio-originator, accepting that it will never be possible to manufacture a product that is identical, in part owing to microheterogeneity. The biosimilar product must also resemble the bio-originator in terms of in vitro assays ranging from antigen binding to Fcγ receptor binding and complement activation.
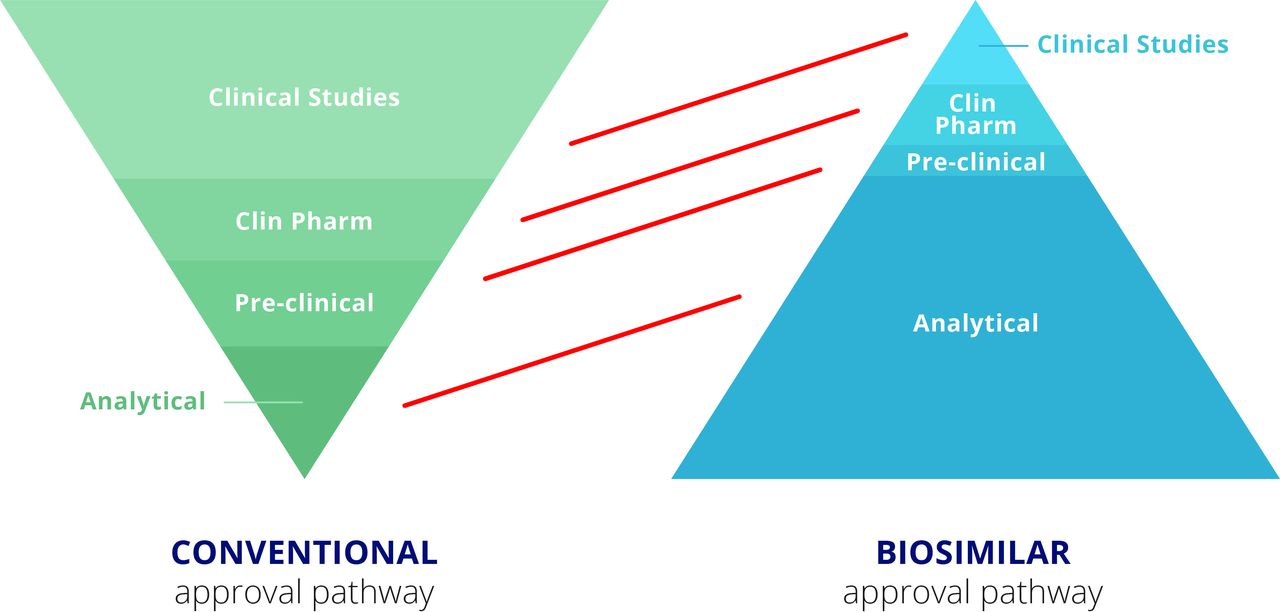
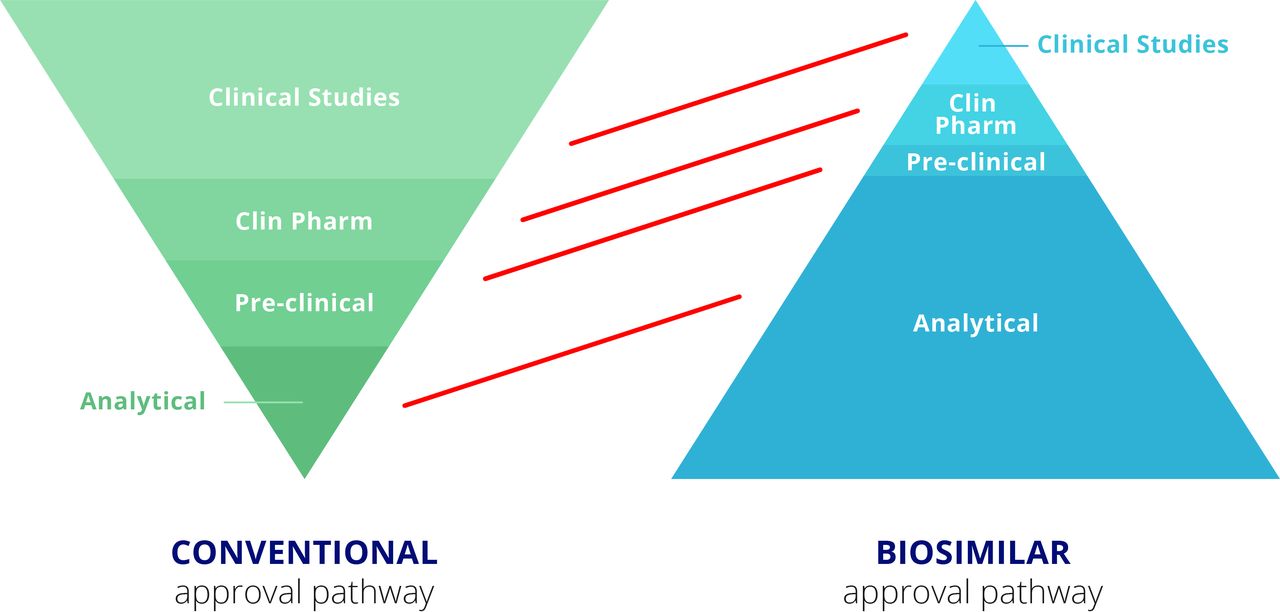
Thus, where the focus for the manufacturer of the bio-originator is to clearly demonstrate safety and efficacy of the therapeutic in clinical trials, the pathway for the manufacturer of the biosimilar is quite different (figure 3).7 The bio-originator manufacturer must produce an analytical package for the regulators that specifies the drug’s composition and formula. Preclinical testing must be undertaken to identify potential toxicities and, where relevant, demonstrate therapeutic effects in an animal model. However, the major work and costs for a bio-originator manufacturer relate to clinical trials. These range from phase I clinical pharmacology studies to clearly demonstrate the pharmacokinetics of the drug, through extensive phase II and III efficacy and safety trials to demonstrate unequivocal therapeutic benefit without excess toxicity. For the biosimilar, in contrast, therapeutic benefit is already identified. Thus, most of the work for the biosimilar manufacturer relates to the analytical package, confirming that the product looks highly similar to the bio-originator in terms of its physicochemical characteristics as well as in terms of its composition, including impurities and aggregates, for example. In vitro activity must also match that of the bio-originator in a wide range of relevant assays. The steps in the biosimilar process are described in more detail below.
{kind=link}
{kind=link}
{kind=link}
Regulatory requirements for approval of a conventional biologic and a biosimilar.
Extensive physicochemical and biological characterisation
To meet regulators’ requirements, the biosimilar manufacturer must first show that the biosimilar is identical to the bio-originator in terms of primary structure, a straightforward task. More challenging is demonstrating identity in terms of higher order (secondary, tertiary, and quaternary) structures. Enzymatic post-translational modifications resulting from glycosylation must be highly similar if not identical within the limits of testing, and other potential variants resulting from deamidation and oxidation also must be characterised. Impurities and aggregates must also be analysed, without significant differences between biosimilar and bio-originator. These are the critical quality attributes of a biological drug that can affect clinical safety and efficacy. The analytical requirements for biosimilars have in some situations catalysed technical advances, with some bio-originators becoming more highly characterised than previously required.
Comparative in vitro non-clinical studies
Comparative in vitro non-clinical studies are needed to confirm that the biosimilar matches the bio-originator in terms of antigen binding affinity and avidity; binding to the Fc receptor for IgG (Fcγ receptor), neonatal Fc receptor, and complement components; functions associated with the antigen binding (Fab) fragment, such as neutralisation and receptor activation or blockade; and functions associated with the fragment crystallisable region (Fc), such as antibody dependent, cell mediated cytotoxicity, complement dependent cytotoxicity, and apoptosis of target cells.
In vivo non-clinical studies
Non-clinical studies in animals may not be required unless there are clear differences between the biosimilar and bio-originator in terms of process related impurities or important differences in formulation that might cause potential efficacy or toxicity concerns. Even then, an informative animal model must be available for such studies to take place.
Randomised controlled trials
Randomised controlled trials (RCTs) for biosimilars aim to reduce any residual uncertainty following the extensive physicochemical and in vitro analyses referred to above. The concept here is that if all physicochemical and in vitro data suggest that biosimilar and bio-originator are equivalent, then clinical benefit can largely be assumed.8 In this way, biosimilars require a single pharmacokinetic study to demonstrate equivalence with the bio-originator and one sufficiently large RCT to demonstrate clinical equivalence alongside comparable safety and immunogenicity. The equivalence margins for this RCT are derived from a meta-analysis of placebo controlled phase III RCTs of the bio-originator.
The study populations for the pharmacokinetic and efficacy studies should represent indications for which approval is being sought. For a drug such as infliximab, for example, patient populations might include rheumatoid arthritis and ankylosing spondylitis. However, using similar arguments to those that require modest clinical testing following demonstration of physicochemical and in vitro equivalence, regulators generally allow extrapolation between indications if efficacy relies on a similar mechanism of action in each indication and equivalence has been demonstrated in one of them.8 Thus, a biosimilar tumour necrosis factor inhibitor may be approved in rheumatoid arthritis, psoriasis, ankylosing spondylitis, psoriatic arthritis, and Crohn’s disease without the need for clinical trials in each condition, provided the bio-originator was licensed for all of them and equivalence between biosimilar and bio-originator was demonstrated in at least one.
Immunogenicity is a major determinant of secondary loss of efficacy for biological drugs. It can be influenced by many factors including route of administration, dosing schedule, disease being treated, patient’s genetic profile, concomitantly prescribed drugs, aggregates in the preparation, and post-translational modifications. The precise effect of post-translational modifications, such as glycosylation, on immunogenicity cannot be predicted. Nonetheless, regulators require immunogenicity to be measured in biosimilar clinical trials and to be kept under review after launch, to monitor for differences that may become apparent with increased exposure of patients during real life use of the biosimilar.
Differences between trials for biologics and biosimilars
The design of trials for biosimilars, even for biosimilars of the same bio-originator, varies considerably. If trials were standardised in various aspects (Box),8 making indirect comparisons between different biosimilars would be easier.
It is worth emphasising that, if a manufacturer is developing a biosimilar for licensing in Europe, all the licensing studies must be conducted with the bio-originator product that is used in Europe. If this is not the case, a bridging study would then be required to demonstrate equivalence between the bio-originator product used and that licensed in Europe, incurring significant additional costs.
Aspects of clinical trial design for biosimilars that could potentially be standardised
Healthy participants versus patients (in phase I)
Inclusion and exclusion criteria
Equivalence margins
Primary endpoint (including timing of assessment)
Secondary endpoints (including timing of assessment)
Pharmacokinetic assays (endpoints compared and timing of assessment)
Immunogenicity (assays used and timing of testing)
Analysis of effects of immunogenicity on pharmacokinetics, efficacy, and safety
Definition of adverse events, such as injection site reactions
Statistical analyses
Crossover designs beyond primary endpoint (in phase III)
Considering the implications for biosimilars
The most obvious advantage of biosimilars is cost. They are cheaper to produce than bio-originators, in part because of likely improvements and increased efficiency of the production process since the bio-originator product was first manufactured. However, the bulk of the cost saving derives from the abbreviated clinical trial programme for biosimilars. Furthermore, as a biosimilar is a copy of an effective drug, the risk of failure is much lower for a biosimilar than its bio-originator. The reduced costs will, hopefully, broaden the availability of biologics to more patients.
Another less obvious advantage of biosimilars is that they are much better characterised than the bio-originator products owing to the rigorous analytical process required for regulatory approval. This means that we better understand what we are injecting into our patients.
The key disadvantage of biosimilars is the possibility of unexpected differences compared with the bio-originator due to, for example, post-translational modifications that happen to influence key functions. However, the regulatory pathways have been designed specifically to minimise this risk.
Registries and post-marketing surveillance
The reduced clinical trial programme means that biosimilars are licensed after relatively few patients have been exposed—often several hundreds rather than several thousands—which means that rare adverse events may be missed. Furthermore, data on long term safety and efficacy may not be available, as long term extension trial data are not routinely required for biosimilar drugs. Registries and post-marketing surveillance studies are consequently all the more critical.
Switching, substitution, and interchangeability
Clinicians frequently switch patients from a branded drug to a generic, using the international non-proprietary name (INN) so the pharmacist may prescribe the drug that is cheapest at a given time. The situation is clearly more complicated for biosimilars, which are never identical to bio-originators, and prescribing is usually by brand name. Thus, whereas clinicians may be comfortable switching from a bio-originator after some experience with a particular biosimilar, they are unlikely to be happy for such switching to occur in the pharmacy without their knowledge. Furthermore, there may be concern around switching to a newer, cheaper biosimilar; this concern may in part be allayed by standardisation of trial designs as mentioned above.
Substitution of biosimilars by pharmacists when a clinician has intended a bio-originator to be prescribed remains a highly controversial issue. The US Food and Drug Administration has proposed trials to identify ‘interchangeable’ products that do not result in immunogenicity despite multiple (at least three) switches between biosimilar and bio-originator; such a biosimilar may then become an interchangeable product, with switching (substitution) permitted at the pharmacy level. At present, no biosimilar has gained this status.
Naming of biosimilars
How to name biosimilars is also somewhat contentious. Infliximab and rituximab are INNs. In the US, the INN of a biosimilar is not the same as the INN of the originator, with a suffix usually used to denote that a product is a biosimilar. In Europe, bio-originators and biosimilars have the same INN and are therefore prescribed by brand name to distinguish between the bio-originator and biosimilar.
Manufacturing changes
Manufacturing processes have changed considerably over the past 20 years to become more efficient, and production locations may have changed, both of which can lead to variation between batches. In 2010 a change in the manufacturing process for Enbrel (etanercept bio-originator) meant that two batches with different glycosylation profiles were available simultaneously.9 Similar changes have been demonstrated for rituximab, with potential implications for therapeutic cytotoxicity.9 The manufacturing process for Remicade (infliximab bio-originator) has undergone more than 35 changes during its lifecycle, so the product given to patients today is not identical to that originally prescribed. Regulators require comparability exercises between old and new batches to confirm that there is no variation that is likely to affect efficacy or safety; invariably, such comparisons suggest the manufacturing changes are unlikely to alter the clinical profile, and label changes are not required.9 10
Intended copies
Intended copies are copies of bio-originators that have not undergone the stringent regulatory process for biosimilars. They are not available in highly regulated markets such as the US, Europe, and Australia but are marketed in less affluent countries.11 Reditux is an example of an intended copy of rituximab available in India, where it has undergone a phase III trial to confirm that it is effective but has not been subject to a head-to-head comparison with originator rituximab. Intended copies are cheap and have broadened accessibility of biologics in less regulated countries, but they are not without problems. Kikuzubam, another intended copy of rituximab that was available in some countries in South America, was withdrawn because of side effects and toxicities.
Considering the approval process – A SUMMARY
Growth factors, hormones, soluble receptors, and monoclonal antibodies are complex biological therapeutics. Biosimilars cannot be identical to their bio-originators owing to their production from living cells. In theory, subtle differences could result in unexpected consequences in terms of safety and efficacy, but the stringent regulatory process has been designed to minimise the likelihood of such an occurrence. A truncated clinical development programme and consequent reduced exposure of patients at the time of marketing necessitate extra vigilance in terms of post-marketing surveillance and registries. Controversial areas remain, including extrapolation of indications and substitution of biosimilars by pharmacists. It is worth emphasising that well documented changes in bio-originator preparations, due to manufacturing changes and plant relocation, reassuringly have not changed long term safety or efficacy profiles.